
Diffuse Cystic Lung Diseases: Definitive Diagnosis is Essential for Targeted Treatments
Diffuse cystic lung diseases (DCLDs) are uncommon and frequently misdiagnosed, in part because there are so many different causes. The most frequently encountered DCLDs in clinical practice are lymphangioleiomyomatosis (LAM), Birt-Hogg-Dubé syndrome (BHD), and pulmonary Langerhans cell histiocytosis (PLCH).1 But cystic lung disease can also be due to Light Chain Deposition Disease or a rare manifestation of follicular bronchiolitis complicating connective tissue disorders such as Systemic Lupus Erythematosus and Sjögren’s Syndrome.2 To raise the profile of DCLDs in the pulmonary community and demystify the diagnostic approach, Francis X. McCormack, MD, Director, Division of Pulmonary, Critical Care & Sleep Medicine at the University of Cincinnati Medical Center and Nishant Gupta, MD, Assistant Professor, Department of Internal Medicine at UC Medical Center recently co-authored a comprehensive two-part review in the American Journal of Respiratory and Critical Care Medicine.3,4 In this interview, the two physicians discussed the primary challenges, pathways to correct diagnosis, and recent scientific developments for the DCLDs.

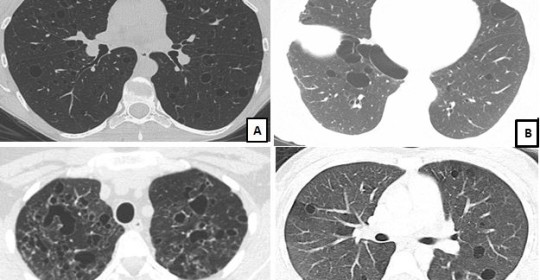
Key to Images/Quiz: A: LAM: CT chest showing round, uniform, thin-walled cysts present diffusely with a normal appearing intervening lung parenchyma B: BHD: CT chest showing oval, lentiform cysts with thin walls, predominantly in the basilar regions, and abutting the pleural surface C: PLCH: CT chest showing multiple bizarre shaped cysts of varying sizes, present in an upper lobe predominant distribution D: Sjogren’s syndrome related cystic lung disease: CT chest showing multiple round-oval cysts in a random distribution. Note, that some of these cysts have an eccentric vessel, and some cysts also seem to have internal structures within them.
Diagnostic Dilemma
Both clinicians spoke of the difficulty of making an accurate and timely diagnosis due to the rarity of the DCLDs. “These diseases generally get only limited attention during pulmonary fellowship,” said McCormack. In many cases of DCLD, shortness of breath prompts the clinician to order lung function tests and a chest x-ray, which may well yield few clues as these diseases typically present with a non-specific obstructive pattern and are commonly invisible on a simple radiograph. DCLD patients are often labelled as having asthma or chronic obstructive pulmonary disease (COPD), but often don’t fit the standard profile, as a smoking history may be lacking, and exacerbations and remissions may be absent. This can lead to a delay in diagnosis of up to several years. Clinicians are often resistant to the notion of obtaining CT scans early, dissuaded by normal chest films and the small but measurable radiation exposure risk. In other cases, the incidental finding of multiple cysts on CT scans obtained for unrelated purposes can lead to the search for a diagnosis. Even when a CT scan is obtained and scattered cysts are seen, pulmonary physicians may balk at lung biopsy, since that approach is less familiar for evaluation of DCLDs than for the scarring interstitial lung diseases.
Often, says Gupta, patients without a definitive diagnosis will receive several years of unsuccessful treatments before being referred to a specialty center for additional evaluation. Sometimes, a lung rupture or pneumothorax will result in an Emergency Department (ED) visit and provide for renewed focus on the underlying disorder.
Greater Awareness Needed
McCormack says, “Cystic lung disease isn’t a disease category that we’re generally taught in training to consider as a platform for differential diagnosis in the same ways as we are with obstructive lung disease, restrictive lung disease, interstitial lung disease, and pulmonary vascular disease. “This is a fairly new concept,” says McCormack. Gupta elaborates, “There aren’t many effective therapies available for this class of diseases, but for the few that are so blessed, we believe we can actually change the natural history of the disease. Consequently, it is no longer acceptable to postpone or miss the diagnosis.”
Recent Developments
The last two decades have been marked by extraordinary advancements in knowledge regarding cystic lung disease. Gupta cites the example of lymphangioleiomyomatosis (LAM), a rare, progressive, cystic lung disease, pointing out, “Science has progressed from knowing virtually nothing about the disease to understanding its natural history, finding diagnostic and predictive biomarkers and pinpointing a treatment for LAM in less than 20 years.” He continues, “We hope to take the example of LAM and apply it to other cystic lung diseases.”
McCormack cites recent progress in the understanding of pulmonary Langerhans cell histiocytosis (PLCH): “PLCH has some striking similarities to LAM, particularly in that it is caused by DNA mutations that lead to abnormal cell proliferation and invasion of the lung with a destructive result. In PLCH, mutant myeloid cells are recruited to the lung by cigarette smoke and dissolve it. We know something about the genetic mutations that are driving that process, which provides a window into the signaling pathways involved and identifies targets for treatment.”3 In fact, Gupta and McCormack recently submitted an NIH grant to conduct a trial with a highly targeted drug for the mutation that occurs in the cells in patients with PLCH.
Moving Forward
According to Gupta, a key goal for the future is to increase awareness of cystic lung diseases among emergency medicine physicians. He believes that up to 10-15% of the pneumothoraces in the Emergency Room are caused by heritable or sporadic genetic mutations, and that CT scanning of the chest on the first pneumothorax is cost-effective. He and McCormack will soon publish studies supporting this approach.

Network of clinics in Rare Lung Disease Consortium
References:
-
Gupta N, Meraj R, Tanase D, James LE, Seyama K, Lynch DA, et al. Accuracy of chest high-resolution computed tomography in diagnosing diffuse cystic lung diseases. Eur Respir J. 2015.
-
Lohrmann C, Uhl M, Warnatz K, Ghanem N, Kotter E, Schaefer O, et al. High-resolution CT imaging of the lung for patients with primary Sjogren’s syndrome. Eur J Radiol. 2004;52(2):137-43.
-
Gupta N, Vassallo R, Wikenheiser-Brokamp KA, McCormack FX. Diffuse Cystic Lung Disease. Part I. Am J Respir Crit Care Med. 2015;191(12):1354-66.
-
Gupta N, Vassallo R, Wikenheiser-Brokamp KA, McCormack FX. Diffuse Cystic Lung Disease. Part II. Am J Respir Crit Care Med. 2015;192(1):17-29.
 Francis X. McCormack, MD
Francis X. McCormack, MD
Director, Division of Pulmonary, Critical Care & Sleep Medicine
Taylor Professor of Medicine, Department of Internal Medicine
(513) 558-4831/(513) 475-8523
mccormfx@ucmail.uc.edu
Connect with Francis McCormack, MD on Doximity
 Nishant Gupta, MD
Nishant Gupta, MD
Assistant Professor, Department of Internal Medicine
(513) 558-4831
guptans@ucmail.uc.edu
Connect with Nishant Gupta, MD on Doximity
Leave a reply →