
Miracle Drugs for Cystic Fibrosis Are Here, with More to Come
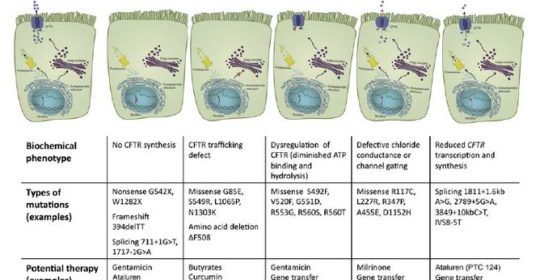
Long-involved in clinical trials for agents to treat cystic fibrosis (CF), Patricia Joseph, MD, professor of medicine and director of Cystic Fibrosis program in the department of internal medicine, University of Cincinnati (UC) Medical Center, says that therapies targeted at different cystic fibrosis (CF) defects are “the most exciting developments in CF right now.” There are five different classes of gene defects in CF, all of which alter the chloride and sodium content of the mucous layer in the airways of the lung. “Because no single CFTR [CF transmembrane conductance regulator]-modulating therapy works for all defects, therapies must be targeted to the specific gene mutation,” says Dr. Joseph.
According to Dr. Joseph, “The modulator drugs are the first to show long-term benefit.” Currently available therapies include potentiator therapy, such as ivacaftor, which corrects the defect in chloride transport and allows the ion channel to open; and corrector drugs, such as lumacaftor-ivacaftor, which help process the CFTR protein and transport it to the cell membrane.
New Drug Agents for Cystic Fibrosis
The recently approved agents, ivacaftor and lumacaftor-ivacaftor, slow CF disease progression, improve their quality of life and may prolong survival. Ivacaftor is approved for the 4% to 5% of CF patients who have the G551D mutation,1 and lumacaftor-ivacaftor is indicated for CF patients with two copies of Phe508del, the most common CF mutation that is present in 45% of CF patients, now numbering over 30,000 individuals in the U.S.2,3
In a double-blind, randomized, placebo-controlled trial of ivacaftor for patients with the G551D mutations, patients in the treatment arm experienced improved lung function at day 15 of the trial, an effect that was maintained throughout the study, as well as improvements in the frequency of pulmonary exacerbations, weight, concentration of sweat chloride, and patient-reported respiratory symptoms.1 The results from two phase 3, randomized, double-blind, placebo-controlled studies of lumacaftor-ivacaftor showed sustained and significant improvements in FEV1, clinically meaningful lessening of pulmonary exacerbations, a consistent increase in body mass index, and improvements of various health-related measures.2 A similar number of patients reported adverse events in the treatment and placebo groups. The FDA has approved ivacaftor for the treatment of patients with the G551D mutations and combination therapy with both lumacaftor and ivacaftor for the treatment of patients with the Phe508del mutation.
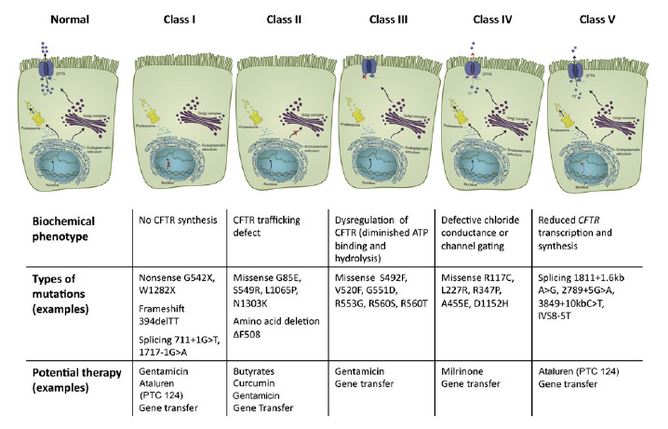
The CFTR mutations have been divided into five classes based on their impact on transporter molecule or regulation.7
Ataluren and Other CFTR modulators on the Horizon
Ataluren, which is now being evaluated for approval by the Food and Drug Administration, is aimed at Class 1 ‘nonsense (premature stop codon)’ mutations that are present in about 10% of CF patients4 and interrupt the synthesis of the CFTR protein. Ataluren promotes ‘read-through’ of the translational complex in which the ribosome ignores the mutant stop codon and synthesizes the protein. Ataluren has already been approved in Europe for patients with Duchenne muscular dystrophy.5 In one CF study, investigators found no significant differences in FEV1 or pulmonary exacerbations between the Ataluren and placebo groups, although a posthoc analysis determined that Ataluren appeared to benefit patients who were not using chronic inhaled tobramycin.4
Companies that are developing and testing CFTR modulators include Nivalis (N9115), Bayer (riociguat), and ProQR (QR 010), and Vertex, says Dr. Joseph. Currently, UC Medical Center is participating in several CF trials: Vertex 661-107, in which the mutations are unlikely to respond, and the trial is paused; Vertex 661-108, a study of mutations with residual function; and Vertex 661-109, a study of gating defects.6 “We are also involved in Vertex 661-110, the open-label extension of the previous three studies. PTC-124 or Ataluren, which is directed to nonsense mutations, is closed except for the open-label extension,” says Dr. Joseph.
The UC Adult CF Program Treatment Approach
Currently, the UC Adult CF Program treats 150 CF patients, including 10 to 12 who are taking ivacaftor, approximately 50 who are candidates for lumacaftor-ivacaftor, and 10 who are participating in studies of other modulators. (Approximately 10 post-transplant patients are not drug candidates.) In general, the ivacaftor patients have had very gratifying responses, especially those who are in the early stages of CF. The response to lumacaftor-ivacaftor has not been as dramatic, says Dr. Joseph, and many patients have contraindications for the combination therapy. Generally, however, most patients are “doing well on the drugs,” says Dr. Joseph.
Dr. Joseph stresses that patients should continue their usual treatment regimen when they begin one of the newer drugs: Some patients initially did “significantly worse” because they dropped vital routine treatments such as airway clearance, inhaled medications, pancreatic enzyme supplements. “Fortunately, they improved after resuming their full treatment regimens.”
Regarding future CF drugs, Dr. Joseph anticipates improved versions of ivacaftor and lumacaftor-ivacaftor, and agents that will treat mutations that are not currently addressed by available therapies. She adds. “It has been a rare privilege to care for CF patients in the era of targeted molecular therapies. I am thrilled for my patients that science has brought so much hope and promise to CF.”
References:
1.Ramsey BW, Davies J, McElvaney G, et al, for the VX08-770-102 Study Group. A CFTR potentiator in patients with cystic fibrosis and the G551Dmutation. N Engl J Med. 2011;365:1663-1672.
2.Wainwright CE, Elborn JS, Ramsey BW, et al, for the TRAFFIC and TRANSPORT Study Groups. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373:220-231.
3.Cystic Fibrosis Foundation. About cystic fibrosis. Available at: www.cff.org/What-is-CF/About-Cystic-Fibrosis. Accessed:March 3, 2016.
4. Kerem E, Konstan MW, De Boeck K, et al, for the Cystic Fibrosis Ataluren Study Group. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomized, double-blind, placebo-controlled phase 3 trial. Lancet Respir Med. 2014;2:539-547.
5. Translarna [ataluren]. European Medicines Agency. Available at: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002720/human_med_001742.jsp&mid=WC0b01ac058001d124. Accessed: March 10, 2016.
6. Current and upcoming CFFT approved clinical trials. Cystic Fibrosis Foundation.Available at: https://www.cff.org/Our-Research/Clinical-Trials/. Accessed:March 10, 2016.
7. Herrmann U, Dockter G, Lammert F. Cystic fibrosis-associated liver disease. Best Practice & Research in Clinical Gastroenterology 2010;24:585-592.
 Patricia Joseph, MD
Patricia Joseph, MD
Professor of Medicine, Department of Internal Medicine
(513) 558-4826
josephpm@ucmail.uc.edu
Connect with Patricia Joseph, MD on Doximity
Leave a reply →